
Vepdegestrant FDA Approval: First PROTAC for Breast Cancer
Executive Summary
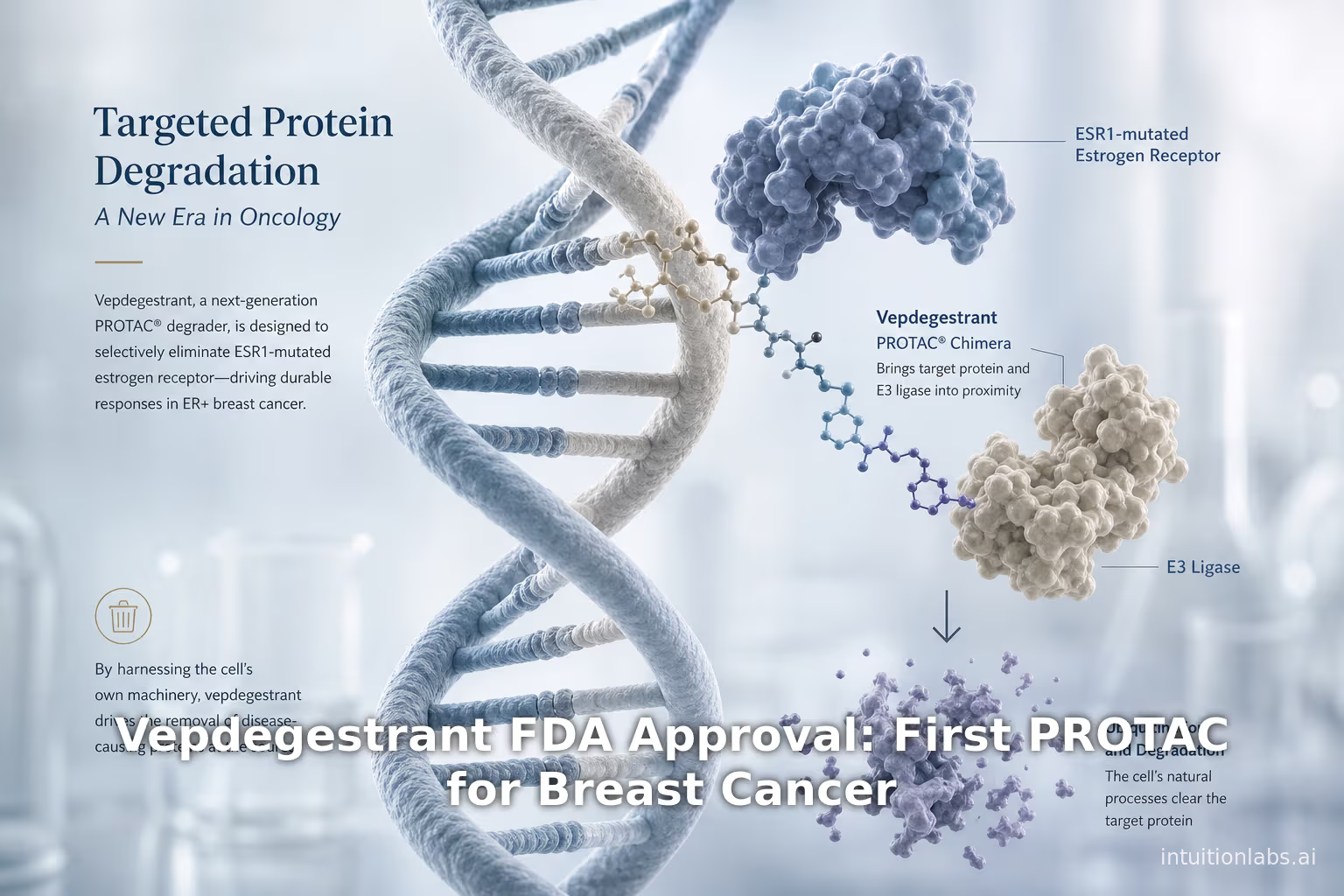
On May 1, 2026 the US Food and Drug Administration (FDA) approved vepdegestrant (brand name Veppanu, Arvinas/Pfizer) for adults with estrogen receptor–positive (ER+), HER2-negative, ESR1-mutated advanced or metastatic breast cancer following progression on prior endocrine therapy ([1]) ([2]). Vepdegestrant is notable as the first-in-class PROTAC (PROteolysis TArgeting Chimera)–based therapy to reach the clinic. In clinical trials (notably the phase 3 VERITAC-2 study), vepdegestrant significantly improved progression-free survival (PFS) over fulvestrant in the ESR1-mutant population – roughly 5.0 vs. 2.1 months median PFS (hazard ratio 0.57, p<0.0001) ([3]) ([4]) – and yielded higher objective response rates (19% vs 4%). The safety profile was generally manageable, with most adverse events (e.g. laboratory abnormalities, fatigue, nausea) being low-grade ([5]) ([6]). Importantly, vepdegestrant is an oral PROTAC that degrades the estrogen receptor protein, “trashing [the receptor] via the cell’s natural waste disposal system” rather than merely inhibiting it ([7]).
Vepdegestrant’s approval validates targeted protein degradation as a viable therapeutic modality in human cancer ([8]) ([9]). It addresses the large unmet need in ER+ breast cancer: up to 40–50% of patients on prior endocrine/CDK4/6 therapy harbor activating ESR1 mutations that confer endocrine resistance ([10]) ([11]). By providing a new oral option for this genetically-defined subset, the approval is already being hailed as a “transformative moment” and “encouraging development” for patients ([8]) ([12]). More broadly, this milestone is expected to accelerate the development and investment in protein degraders: dozens of PROTACs and molecular “glues” are in clinical and preclinical pipelines across oncology, immunology and neurology. Analysts note that 2026 may mark a commercial inflection point for the protein degradation field (“a historic cusp from concept to commercialization” ([8])). Companies like Arvinas and Kymera – the early pioneers – will likely see expanded pipelines, licensing deals, and heightened R&D activity. At the same time, long-term success is not guaranteed, and close attention will be paid to broader safety, efficacy in larger populations, and competition from other modalities. Nonetheless, Veppanu’s approval establishes a regulatory precedent and invigorates the previously mostly-theoretical promise of PROTAC technology.
This report provides a comprehensive overview of the background, clinical data, and implications of this first-of-its-kind approval. We review the history of endocrine therapy and ESR1-mutant breast cancer, the PROTAC mechanism of action, and the pivotal trial data for vepdegestrant. We compare Veppanu to existing therapies (such as fulvestrant and oral SERDs) and place it in context of the emerging targeted protein degradation pipeline. We present relevant data and expert perspectives, including efficacy statistics and case examples, and analyze the likely impact on drug development. Finally, we discuss future directions: new therapeutic opportunities unlocked by protein degradation, regulatory and commercial considerations, and the prospects for a wave of next-generation degrader drugs across multiple disease areas.
1. Introduction and Background
Breast cancer is the most common cancer in women worldwide ([13]). Roughly 70–80% of breast tumors express the estrogen receptor (ER) and are initially driven by estrogen signaling ([14]). Standard first-line treatment for ER-positive/HER2-negative advanced breast cancer (ABC) has traditionally been endocrine therapy (ET): agents that suppress estrogen production (aromatase inhibitors, AIs) or block ER function (selective ER modulators/degraders). For example, tamoxifen (a selective estrogen receptor modulator, SERM), aromatase inhibitors (letrozole, anastrozole, exemestane), and fulvestrant (a selective ER degrader/SERD given by injection) are cornerstone therapies in ER+ disease. These agents act via occupancy-driven pharmacology: the drug binds to the ER and inhibits its activity (Figure 1a).
Over the past 15 years, combinations of ET with CDK4/6 inhibitors have greatly extended control of ER+ metastatic disease. Yet, almost all patients eventually develop endocrine resistance. A key mechanism of acquired resistance is mutation of the ER gene, ESR1, which encodes ERα. ESR1 ligand-binding domain (LBD) mutations (most notably D538G, Y537S, Y537N, Y537C, etc.) stabilize the receptor in an active conformation, making it constitutively active and less sensitive to estrogen deprivation ([14]) ([15]). Clinically, ESR1 mutations occur mainly in advanced/metastatic settings: approximately 20–40% of metastatic ER+ patients treated with aromatase inhibitors harbor ESR1 mutations ([14]). One review notes that roughly 50% of endocrine-resistant metastatic cases involve ESR1 alterations ([16]). ESR1-mutant clones are strongly selected by prior endocrine therapy (aromatase inhibitors or tamoxifen) but are rare (<5%) in treatment-naive or early-stage tumors ([14]) ([17]). In practice, ESR1 mutations are now commonly assayed by circulating tumor DNA (e.g.Guardant360) or tissue sequencing, as they define a subset of patients whose cancers no longer respond well to aromatase inhibitors ([14]) ([18]).
The emergence of ESR1-mutant breast cancer has driven renewed interest in ER-targeting agents. Fulvestrant, an intramuscular SERD that degrades ERα, has been a staple for postmenopausal or metastatic ER+ disease. More recently, new oral SERDs (selective ER degraders) and ER antagonists have shown promise. For example, the FDA approved elacestrant (Orserdu) in January 2023 for ER+/HER2–, ESR1-mutant advanced breast cancer that progressed on prior endocrine therapy ([19]). In the EMBERGLASS-1 trial, elacestrant improved median PFS to 3.8 vs 1.9 months (HR 0.55) in ESR1-mutant patients ([20]). Other emerging ER-targeted therapies include oral SERMs/SERDs from Lilly (giredestrant) and Roche (enersiertib), varying selective estrogen receptor covalent antagonists (SERCAs) and selective estrogen receptor covalent cholesterols (SECAs) under study, and steroid sulfatase inhibitors. Yet even with these advances, second-line options for heavily pretreated patients remain limited. As one expert noted: “For patients living with ESR1 mutant, ER+/HER2– advanced breast cancer, there have been minimal second-line treatment options once standard therapies are no longer effective” ([12]).
Proteolysis-targeting chimeras (PROTACs) represent a fundamentally different strategy. First conceptualized in the early 2000s, PROTACs are heterobifunctional small molecules that simultaneously bind a target protein and recruit an E3 ubiquitin ligase (Figure 1b) ([21]). This brings the ubiquitin-proteasome system to tag and fully degrade the target, instead of merely inhibiting it. In effect, PROTACs enact event-driven pharmacology, catalytically destroying disease proteins ([21]) ([22]). Early demonstrations confirmed the concept: for example, Rodriguez-Gonzalez et al. (2008) designed PROTACs that linked estradiol to a VHL-ligand; these compounds induced proteasomal ERα degradation and G1 arrest in ER-dependent breast cancer cells ([23]). Since then, hundreds of PROTAC programs have emerged, leveraging different E3 ligases (CRBN, VHL, MDM2, IAPs, etc.) and targeting oncogenes, transcription factors, kinases, and more ([24]) ([25]). PROTACs can degrade “undruggable” targets lacking traditional active sites, escape resistance to occupancy inhibitors, and may require lower drug exposure because one molecule can act catalytically ([21]) ([22]). (Bayer, for example, notes that PROTACs do not have to occupy the binding site constantly and may yield longer-lasting effects ([22]).)
To date, no PROTAC had been approved by regulatory agencies. Commercial development was mostly at clinical or preclinical stages. Leading the field, Arvinas (co-founded by Craig Crews and his team) has advanced PROTACs for hormone-driven cancers – notably AR degraders for prostate cancer (ARV-110 and ARV-766) and an ER degrader (ARV-471). In parallel, other biotechs (Kymera, C4 Therapeutics, Nurix, etc.) and large pharmas have built PROTAC and molecular-glue pipelines in oncology, immunology, neurology and eye disease. The FDA’s greenlight of vepdegestrant/Veppanu in 2026 thus marks a turning point: the transition of targeted protein degradation from experimental modality to validated therapeutic platform ([8]) ([7]). The remainder of this report delves into each aspect of this development, from molecular mechanism to clinical evidence to industry impact.
2. ER+ Breast Cancer and ESR1 Mutations
2.1 Hormone-Driven Breast Cancer and Therapies
Estrogen signaling drives proliferation in the majority of breast cancers. Early-stage and metastatic ER-positive (often HER2-negative) breast cancers are generally managed with hormone therapies. Tamoxifen, a SERM, and fulvestrant, a SERD, directly antagonize ER; aromatase inhibitors (AIs, e.g. letrozole, anastrozole, exemestane) lower systemic estrogen. These treatments exploit occupancy-driven pharmacology, binding to the ER ligand-binding domain (LBD) or inhibiting its ligand and thereby blocking ER transcriptional activity ([21]). For many patients, endocrine therapy yields durable responses; however, most eventually acquire resistance.
A potent mechanism of escape is mutation of the ESR1 gene. Characteristic ESR1 mutations (e.g. D538G, Y537S/N/C, etc.) occur exclusively in the LBD and stabilize ER in an activated conformation even without estrogen ([15]) ([16]). These mutations diminish the efficacy of AIs (which can no longer deflate estrogen drive) and, in vitro, reduce sensitivity to tamoxifen or fulvestrant ([26]) ([27]). Clinically, ESR1 mutations are strongly associated with prior exposure to aromatase inhibitors in the metastatic setting ([14]).
Multiple studies of endocrine-resistant metastatic breast cancer report ESR1 mutation prevalence around 30–40%, depending on patient population. For instance, a meta-analysis noted ~20–40% of metastatic patients on an AI harbor an ESR1 mutation ([14]). Among patients who have had one line of endocrine therapy plus a CDK4/6 inhibitor, the rate may reach 40–50% ([10]) ([11]). (By contrast, ESR1 mutations are rare (<5%) in treatment-naïve early-stage disease ([17]).) These statistics are echoed by both academic reviews and industry sources: Arvinas reports that “for those previously treated with endocrine therapy and a CDK4/6 inhibitor for metastatic disease, ESR1m mutations occur in up to 40%–50% of patients” ([10]). In short, ESR1 mutations have become a common driver of resistance in ER+ metastatic breast cancer.
Clinicians now routinely test for ESR1 mutations (often via liquid biopsy) to inform second-line therapy. For example, the FDA approved Guardant360® CDx to identify ESR1-mutant patients eligible for both elacestrant (Orserdu) and vepdegestrant ([28]) ([29]). In practice, an ER+ patient who progresses on first-line ET (with or without CDK4/6 inhibitors) may be sequenced; if ESR1-mutant clones are detected, agents specifically targeting mutant ER become options. Prior to May 2026, the main targeted options included fulvestrant or new oral SERDs like elacestrant ([20]), and experimental therapies under development (e.g. selective ER covalent antagonists). Vepdegestrant (Veppanu) adds a novel weapon – a small molecule that actively degrades the ER protein – to this setting.
2.2 Clinical Characteristics and Unmet Need in ESR1-mutant Patients
Patients with ESR1-mutant ER+ breast cancer face a challenging prognosis. By definition, their tumors have already progressed on standard ET. Observational studies suggest that ESR1 mutations confer faster progression on successive hormonal therapies ([16]). For example, retrospective trials (SoFEA, PALOMA-3) found that patients with ESR1 mutations had shorter PFS on fulvestrant or aromatase inhibitor therapy than those without ([15]) ([30]). The net effect is that third-line or later endocrine therapy alone yields only modest benefit, often measured in a few months.
Before vepdegestrant’s approval, options were limited. Fulvestrant remains a go-to choice after AI failure, but it is injectable (monthly) and typically achieves only transient disease control. The oral SERD elacestrant was approved based on a PFS improvement of 3.8 vs 1.9 months in ESR1-mutant patients ([20]), but overall survival benefit was unclear and uptake has been modest. Other new ER-targeted agents (giredestrant, etc.) may improve outcomes somewhat, but as of 2026 none had been approved in the second-line ESR1-mutant setting. This leaves a substantial safety and efficacy void.
As one clinical leader summarized: “patients [with ESR1 mutations] often experience rapid disease progression and face limited options after first-line therapy… Veppanu offers a new therapeutic option by targeting a key biological driver of resistance” ([11]). Indeed, Arvinas’ Chief medical officer noted that approval provides “a new oral treatment option that showed improved progression-free survival when compared to the current standard of care, fulvestrant” ([31]). In short, ESR1-mutant ER+ breast cancer is a setting of high unmet need, where an effective ER-degrading PROTAC could make a meaningful difference.
3. PROTAC Technology and Vepdegestrant’s Mechanism
3.1 PROTAC Design and Mechanism of Action
PROTACs (PROteolysis TArgeting Chimeras) are bifunctional molecules typically comprised of three parts: a ligand that binds the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a chemical linker joining them. When a PROTAC enters a cell, it simultaneously binds the target protein and an E3 ligase, forming a ternary complex. This proximity causes the E3 to ubiquitinate the target protein, marking it for destruction by the cell’s 26S proteasome ([21]) ([25]) (Figure 1b). In effect, the PROTAC hijacks the cell’s natural protein disposal machinery to remove the disease protein entirely. Pettersson and Crews (2019) describe this as an “event-driven” mode of action, in contrast to conventional “occupancy-driven” pharmacology ([21]).
A key feature of PROTACs is their ability to act catalytically. After degrading one molecule of target, the PROTAC dissociates and can engage another target protein. Thus, a single PROTAC molecule can, in principle, degrade many copies of its target over time ([21]) ([22]). Additionally, because the target is eliminated entirely, all of its functional activities (enzymatic or scaffolding) are abrogated, which may yield effects that simple inhibition cannot achieve ([21]) ([22]). The Bayer PROTAC primer highlights these advantages: PROTACs “do not require to constantly occupy the functional site [of the protein]” and by removing the protein, “all its functions are abrogated,” potentially enhancing therapeutic effects ([32]). Furthermore, degradation requires drug binding only transiently, so PROTACs might be used at lower effective doses than conventional inhibitors ([33]).
The concept dates back two decades. The first reported PROTAC was published in 2001: a peptide-based chimera that recruited the SCF^β-TrCP E3 ligase to degrade MetAP-2 ([24]). Early PROTACs were often large peptides tethered to small molecules. A decisive breakthrough came in 2008 when Crews and colleagues created the first all-small-molecule PROTACs using nutlin (an MDM2 ligand) to recruit ubiquitination ([34]). By 2010s, medicinal chemists had discovered small-molecule ligands for key E3s like cereblon (CRBN) and VHL, enabling robust drug-like PROTACs. Numerous academic and industry groups have since accelerated PROTAC research ([34]). A recent review by Pettersson and Crews (2019) reports that PROTACs have now been applied to dozens of targets, including kinases, transcription factors, and scaffold proteins ([35]) ([36]).
Vepdegestrant itself follows this paradigm. It is composed of an ER-binding moiety linked to an E3-recruiting ligand. Arvinas describes vepdegestrant as an “investigational orally bioavailable PROTAC estrogen receptor degrader” ([37]). The drug binds to ERα (both wild-type and mutant forms) and recruits an E3 ligase such as the von Hippel–Lindau (VHL) complex, triggering ubiquitination and proteasomal degradation of ERα. By contrast with fulvestrant (which binds ER and partially destabilizes it), vepdegestrant actively shuttles the receptor to the proteasome. In essence, it “takes unwanted proteins (in this case, estrogen receptors)… and trashes them via the cell’s natural waste disposal system” ([7]). This catalytic degradation effect is at the heart of the PROTAC strategy’s promise.
3.2 Rationale for PROTAC in ER+ Disease
Given the biology above, why might a PROTAC like vepdegestrant succeed where prior drugs have limited effect? First, it may degrade ERα more completely and durably than STRDs or SERMs. In preclinical models, PROTAC ER degraders often show low nanomolar potency (high DC_50) and near-complete receptor knockdown in tumor cells ([38]). This could translate clinically into deeper blockade of estrogen signaling. Second, because PROTACs operate catalytically, they can overcome stoichiometric limitations and potentially degrade mutant ER that is present at high levels or has reduced drug affinity. Notably, early clinical data on ARV-471 (vepdegestrant) suggested activity even against the tough Y537S ESR1 mutation, which is highly resistant to fulvestrant ([15]) ([39]). Third, PROTACs may have distinct resistance profiles. By recruiting a specific E3, resistance would likely require either loss of that E3 or mutation of PROTAC binding, both less common than ESR1 point mutations. Finally, PROTACs open new chemical space: they can target proteins once considered “undruggable” by conventional binding (although ER was druggable, this illustrates the general point) ([21]) ([25]).
Taken together, the development of vepdegestrant leveraged this unique PROTAC mechanism against a well-validated target (ERα) in a population (ESR1-mutant metastatic BC) known to be driven by the target. As Arvinas CEO Randy Teel remarked at approval, vepdegestrant is “based on the technology we’ve pioneered since 2013” and proves that “targeted protein degradation can translate into meaningful clinical impact” ([8]). It thus represents both “deep science” (protein ubiquitination) and a novel drug category in oncology.
4. Vepdegestrant (ARV-471/Veppanu): Clinical Development and Trial Results
4.1 Collaboration and Development Pathway
Vepdegestrant was discovered by Arvinas and developed jointly with partner Pfizer. Arvinas led early research and clinical studies of ARV-471 (the code name for vepdegestrant) starting in phase 1, and after encouraging phase 2 data it advanced to confirmatory trials. ([40]) ([37]) The pivotal study was VERITAC-2 (NCT05654623), a randomized, open-label phase 3 trial of vepdegestrant versus fulvestrant in patients with ER+/HER2- ABC who had progressed after 1–2 prior lines of endocrine therapy (including one with a CDK4/6 inhibitor) ([41]). Enrollment totaled 624 patients, of whom 270 (43%) had ESR1 mutations detected by circulating tumor DNA ([3]). Randomization was 1:1 to oral vepdegestrant 200 mg daily versus intramuscular fulvestrant (500 mg on Days 1 and 15 of cycle 1, then monthly) ([42]). Stratification factors included ESR1 mutation status and presence of visceral metastases ([42]).
The primary endpoint was blinded independent review–assessed progression-free survival (PFS) in the ESR1-mutant population, with secondary analyses of PFS in the overall intent-to-treat cohort, overall survival (OS), and response rates ([41]). According to press reports, the trial “achieved its primary endpoint in the estrogen receptor 1-mutant (ESR1m) population,” with vepdegestrant demonstrating a “statistically significant and clinically meaningful improvement in PFS” compared to fulvestrant ([43]). The ITT population (including both ESR1-mutant and wild-type) did not reach statistical significance, consistent with the expectation that ESR1-mutant patients derive the most benefit.
4.2 Efficacy Results
The VERITAC-2 outcomes in the ESR1-mutant cohort were striking. The median PFS in this group was 5.0 months on vepdegestrant versus 2.1 months on fulvestrant ([3]) ([4]). This corresponds to a hazard ratio of 0.57 (95% confidence interval 0.42–0.77, p<0.001), meaning a 43% reduction in the risk of progression or death ([3]) ([4]). Objective response rate (ORR) was also higher: 19% on vepdegestrant vs 4% with fulvestrant ([3]). These data indicate that vepdegestrant roughly doubled to tripled the time before progression in ESR1-mutant patients compared to standard fulvestrant. It is notable that the control arm’s PFS (2.1 months) aligns with the very limited benefit expected for endocrine therapy in heavily pretreated, ESR1-mutant disease.
By contrast, in the overall ITT population (including ESR1 wild-type), the PFS curves separated less markedly and did not achieve formal significance. Only in the ESR1-mutant subgroup was a clear benefit observed ([43]). This finding is unsurprising given the biology: wild-type ESR1 tumors generally remain more hormone-sensitive, whereas mutated tumors are the ones needing novel blockade. As Arvinas executives noted, over 40% of third-line patients have ESR1 mutations ([10]), so the approval specifically targets that high-need subgroup.
Overall survival (OS) data were still immature at approval. At the time of analysis, only about 16% of ESR1-mutant patients had died ([44]). Therefore, any OS difference could not yet be reliably determined. This is common in oncology approvals based on PFS gains. Ongoing follow-up is needed to see if vepdegestrant extends life, but PFS improvement alone – especially in a resistant setting – is considered clinically meaningful by oncologists.
4.3 Safety and Tolerability
Vepdegestrant’s safety profile in VERITAC-2 appears manageable. According to the FDA and company reports, most adverse effects were low-grade (grade 1–2) ([5]). In the trial, the most common adverse reactions (occurring in ≥10% of patients) included hematologic laboratory changes (decreased white blood cells, neutrophils, and hemoglobin), elevated liver enzymes (AST/ALT, alkaline phosphatase), fatigue, musculoskeletal pain, nausea, decreased appetite, electrolyte changes (low potassium), and QT interval prolongation on ECG ([6]). In other words, vepdegestrant can cause mild bone marrow suppression (neutropenia/anemia), some liver enzyme elevations, and minor GI and systemic symptoms. These were generally tolerable; only a small minority of patients required dose interruption.
Importantly, the FDA label includes specific warnings for QTc prolongation and embryo-fetal toxicity ([45]). Prolonged QT interval was observed in some patients, so ECG monitoring may be warranted. As a contraceptive precaution, women of childbearing potential should avoid pregnancy during treatment and for two weeks after the last dose. These safety issues are standard for a potent endocrine agent. No unexpected safety signals emerged beyond these known class effects. Notably, vepdegestrant is given orally once daily with food (200 mg), which is a convenience advantage over fulvestrant’s intramuscular injections ([46]).
In summary, vepdegestrant’s efficacy in ESR1-mutant patients was moderate but statistically significant (median PFS improved by ~3 months; HR 0.57) ([3]) ([4]), and its toxicity was acceptable with mostly mild lab abnormalities and controlled side effects ([5]) ([6]). FDA reviewers concluded that the benefit–risk ratio was favorable in the targeted population. The Medicinal Products Advisory Committee of FDA (if convened) would have noted that this gave a meaningful new option for a group with few alternatives. As one investigator stated, “the introduction of a new, targeted treatment is encouraging… [it] highlights meaningful innovation” ([12]).
4.4 Comparison with Other Endocrine Degraders
It is instructive to compare vepdegestrant with recent ER-targeting drugs. Fulvestrant (500 mg IM) has been used for decades as a “standard” SERD. In ESR1-mutant metastatic patients, fulvestrant alone typically yields a median PFS <3 months, as seen in VERITAC-2 and in previous trials. In contrast, elacestrant (Orserdu) is a newer oral SERD approved in 2023 specifically for ESR1-mutant advanced breast cancer ([19]). In the phase 3 EMERALD trial, elacestrant achieved median PFS 3.8 vs. 1.9 months (hazard ratio 0.55) in the ESR1-mutant subgroup ([20]). This is roughly comparable to vepdegestrant’s 5.0 vs 2.1 (HR 0.57) in VERITAC-2. Thus, both oral agents approximately doubled to tripled PFS over fulvestrant in this patient subset. The slightly longer PFS with vepdegestrant (5.0 vs 3.8) may reflect cross-trial differences, patient selection, or drug potency; both results, however, confirm ESR1-mutant patients derive a few extra months of disease control with a potent degrader.
An important practical distinction is route and convenience. Fulvestrant requires bi-monthly intramuscular shots and has poor bioavailability, whereas elacestrant and vepdegestrant are oral. Elacestrant’s ORR in ESR1-mutant patients was ~19% ([20]) (similar to vepdegestrant’s 19%), suggesting comparable activity. Another emerging player is giredestrant (a Roche SERD), which in combination with everolimus showed significant PFS benefit over the standard endocrine/everolimus doublet ([47]). (In the Phase 3 EvERA trial, giredestrant plus everolimus significantly improved PFS in both ITT and ESR1-mutant subgroups ([47]).) However, giredestrant is being developed for combination use rather than monotherapy.
In summary, vepdegestrant stands among the most effective ER degraders now entering practice, with efficacy in the same ballpark as elacestrant but with the novelty of a proteolysis-targeting mechanism. Its key advantage over fulvestrant is both its greater activity and oral dosing. Relative to elacestrant, the open question is whether complete protein degradation (via PROTAC) offers any additional benefit; the clinical data so far show similar improvements. Further real-world experience will clarify if any differences (in depth of response, side effects, or outcomes) emerge between PROTAC vs. SERD strategies.
5. Implications for the Protein Degrader Pipeline
The approval of vepdegestrant has broad implications for the entire targeted protein degradation field. We analyze its impact from several angles:
5.1 Validation of the PROTAC Modality
Veppanu’s approval provides “proof of concept” that targeted protein degraders can succeed as medicines. As noted by BioPharma Dive, this is “the first of its kind to get to market”, and vepdegestrant is the “first PROTAC degrader to demonstrate clinical benefit in a Phase 3 trial” ([48]) ([43]). Arvinas emphasized that FDA clearance of Veppanu represents “the first-ever approved PROTAC therapy” ([8]). Regulatory acknowledgement means pharmacologists, investors, and drug developers now have a precedent. The approval establishes a regulatory pathway for this modality, clarifying endpoints and expectations for future PROTAC candidates.
On a strategic level, this success underscores the scientific viability of PROTACs. As one analyst put it, Gilead’s collaboration on PROTACs with Kymera (announced June 2025) “validates [protein degrader] development capabilities” and the notion that degraders “offer a better targeting approach than small molecule inhibitors” ([49]). Veppanu’s performance – 43% risk reduction in PFS – demonstrates a clear clinical effect of protein degradation beyond historical controls. It also shows that drug-like PROTACs can be optimized for oral bioavailability and tolerability in humans. These lessons will feed into ongoing programs. Any hesitancy about PROTAC pharmacology should be allayed by Arvinas’s data.
5.2 Commercial and Competitive Ramifications
Arvinas and Pfizer’s first-in-class approval will influence the business landscape. Arvinas has announced plans to out-license or co-commercialize Veppanu, having “selected a third party” to help bring the drug to market ([50]) ([8]). This indicates that large pharmas see value in partnering for PROTAC commercialization. Indeed, Arvinas’s collaboration with Pfizer (and its 2024 licensing of another PROTAC to Novartis) shows major drug companies are investing heavily in this space ([51]). News of milestone payments and upfront fees underscores this trend. For example, Kymera is receiving up to $85M upfront from Gilead to work on molecular glue degraders ([52]), and up to $975M in future milestones from Sanofi on an IRAK4 degrader ([53]). Such deals imply big pharma confidence that protein degraders (PROTACs and glues) will yield blockbuster results.
By contrast, the Biopharma Dive article cautions that Uncertainties remain around Veppanu’s market uptake ([54]). Veppanu was approved specifically for the ESR1-mutant subgroup, which limits patient numbers. Investors will watch whether clinicians adopt it widely or reserve it for niche use. Analysts doubtless will compare its real-world performance and pricing vs earlier endocrine drugs. Nonetheless, the FDA’s validation is likely to spur waves of research and development. Within days of the approval announcement, industry analysts noted 2026 as a watershed year (“商业化元年”) for protein degraders ([55]). There are already ~390 protein degrader molecules in global development (per one Chinese report) covering oncology and beyond ([55]). We expect that number to grow rapidly.
Key near-term milestones include other firsts: targets beyond ER and other modalities. For example, in oncology PROTACs targeting the androgen receptor (AR) are far along: Arvinas’s ARV-110 for castration-resistant prostate cancer has progressed to Phase 3 and ARV-766 (luxdegalutamide) is in Phase 2 under a Novartis license ([51]). Such AR degraders will be watched closely. In non-oncology areas, the pipeline includes “undruggable” targets like LRRK2 for Parkinson’s (Arvinas ARV-102, Phase 1) and kinases or transcription factors in immunology (Kymera’s IRAK4, STAT6, IRF5 programs ([56]) ([57])). Even in hematology, molecular glues like C4 Therapeutics’ IKZF degrader (cemsidomide) are in Phase 2 for multiple myeloma ([58]).
The success of vepdegestrant may also influence traditional drug categories. Established companies with small-molecule libraries may revisit previous “undruggable” targets using PROTAC strategies. Some may even attempt to convert existing inhibitors into degrader chemotypes. Companies renowned for antibody–drug conjugates (ADCs) and kinase inhibitors are now setting up PROTAC programs to complement their portfolios. For example, Bayer’s informational page cavalierly describes PROTACs as expanding the “druggable space” beyond what traditional inhibitors can achieve ([22]), signaling broad interest.
5.3 Impact on Clinical Practice and Guidelines
In the clinic, Veppanu’s approval will change first-line and later-line algorithms for ER+ metastatic breast cancer. For any patient with progressing ER+ disease, physicians now should test ESR1 status if not already done. An ESR1 mutation will make a patient eligible for veppdegestrant. Given its oral dosing and modest efficacy advantage, many oncologists may favor it over fulvestrant for the ESR1-mutant subgroup. The Arvinas press release highlights that Veppanu addresses a “significant unmet need” ([11]) and provides “another tool in the breast cancer treatment arsenal” ([12]). Clinical guidelines (ASCO, NCCN) will rapidly incorporate PROTACs into their algorithms for ER+ disease. Moreover, this approval legitimizes ESR1 mutation testing as mandatory for metastatic patients, which will have been elevated by the companion diagnosis requirement.
Commercially, insurance coverage and pricing will become important issues. Vepdegestrant is novel and potentially costly; payers will scrutinize its benefit relative to cost and to existing SERDs. If Veppanu’s list price is high, its PFS gain of ~3 months (in trials) may raise debates over incremental value. However, at this early stage the focus is likely on ensuring patient access rather than immediate generic competition.
5.4 Future Directions in Targeted Degradation
Looking forward, vepdegestrant’s approval is only the opening act. The protein degrader pipeline is diverse and accelerating. We highlight several case studies and examples of programs that stand out:
-
AR-Degraders (Prostate Cancer): As noted, Arvinas has advanced ARV-110 (oral PROTAC against AR splice variants) and ARV-766 (“luxdegalutamide,” an AR PROTAC licensed to Novartis) for metastatic castration-resistant prostate cancer ([51]). Promising early data from ARV-110 and ARV-766 had already fueled partnerships; their later-phase results will further validate (or test) the PROTAC paradigm in prostate cancer. These agents target a similarly well-validated hormone receptor. A successful PROTAC in prostate cancer could be another blockbuster. If either obtains approval around 2027–28, it would mark a second class breakthrough (and Arvinas notes that licensor Novartis plans multiple PROTAC oncology trials ([51])).
-
Immunology Programs (Kymera Therapeutics): Kymera, co-founded by Ira Rothenberg, has pioneered PROTACs for immune targets and molecular glue approaches. Its pipeline includes oral PROTACs against STAT6 (KT-621) for eosinophilic/allergic diseases and IRF5 (KT-579) for lupus ([59]) ([60]). The company also has progressed IRAK4 degraders: KT-474 completed Phase 1 and KT-485 has entered partnership-driven trials ([61]) ([53]). Notably, Kymera’s approach extends beyond PROTACs: it is also developing molecular glues, which are conceptually similar degraders that induce ternary complex formation without linkers. For example, KT-200 is described as a “CDK2 molecular glue” to degrade the cyclin-dependent kinase 2 in CCNE1-amplified cancers ([62]). In June 2025, Kymera announced collaborations with Gilead on new molecular glues (targeting CDK2 in breast cancer) ([63]). An analyst commented that these deals “validate…development capabilities” of protein degraders and their promise to target proteins better than classical inhibitors ([49]).
-
Hematologic Programs (C4 Therapeutics): C4 is advancing PROTAC degraders for blood cancers. Its “TORPEDO” platform generates monofunctional and bifunctional degraders (MonoDAC and BiDAC). The lead candidate is cemsidomide (CFT8634), an IKZF1/3 degrader (an analog of immunomodulatory drugs) now in Phase 2 trials for relapsed multiple myeloma ([58]). In 2026, C4 plans pivotal trials (MOMENTUM) for cemsidomide ([64]). Success here would establish PROTACs in hematologic oncology and challenge existing IMiD therapies. C4 also has PROTACs targeting CDK9 and others in preclinical stages.
-
Neurology (LRRK2, Tau, Huntington): Arvinas is testing neurodegraders. ARV-102 is a brain-penetrant LRRK2 PROTAC for Parkinson’s disease and related tauopathy (progressive supranuclear palsy); it began first-in-human studies in 2024 ([65]). Early data at neurological conferences (2025) showed central LRRK2 engagement ([66]). If this yields clinical benefit, it would open PROTACs to neurodegeneration – a completely different indication. Similarly, Arvinas is developing ARV-027, a PROTAC against mutant polyglutamine-expanded androgen receptor for spinal-bulbar muscular atrophy (SBMA/Kennedy’s disease) ([67]). In rodent models, ARV-027 stopped muscle atrophy ([67]). These illustrate the broad reach of degradation technology.
-
Rasopathies and Others: Targeting RAS oncogenes has been a longstanding challenge. Arvinas is pursuing KRAS^G12D PROTACs (ARV-806) in preclinical work ([68]). If successful, this could inaugurate new strategies against RAS-driven cancers. Other groups (Revolution Medicines, RASolute) are exploring molecular glues for Ras as well ([69]). Beyond Ras, companies like Nurix Therapeutics are developing degraders for resistance mechanisms in chronic lymphocytic leukemia (e.g. BTK PROTAC NX-5948) ([70]), and other biotech (Monte Rosa, ImmunoGen, etc.) are applying degraders in immuno-oncology and beyond.
These examples underscore a theme: protein degradation is rapidly expanding into many disease areas. Unlike small-molecule inhibitors limited by the need for a druggable active site, PROTACs/glues can in principle tackle transcription factors, scaffolding proteins, or mutant enzymes. The FDA approval of a PROTAC validates those scientific strategies. It also signals to investors and biotechs that a long lead time (20+ years of research) can pay off, attracting more funding. One industry report even forecasts that 2026 will be the “commercialization year” for this field, with Arvinas’s ARV-471 (vepdegestrant) and upcoming glues marking an inflection point ([55]).
5.5 Tables: Pipeline and Efficacy Examples
To illustrate the landscape, Table 1 lists selected endocrine and PROTAC therapies relevant to ER+ breast cancer. Table 2 summarizes key outcomes from the VERITAC-2 trial (vepdegestrant vs. fulvestrant) in ESR1-mutant patients. These tables are not exhaustive but highlight representative data and comparisons.
Table 1. Selected ER-targeting Therapies in Metastatic Breast Cancer
| Therapy | Modality/Mechanism | Indication (Population) | Administration & Status |
|---|---|---|---|
| Fulvestrant (Faslodex) | SERD (selective ER degrader) | ER+, HER2– metastatic BC after ET | Intramuscular injection, approved (1997) |
| Elacestrant (Orserdu) | Oral SERD | ER+, HER2–, ESR1-mutated advanced BC after ≥1 ET | Oral tablet, approved Jan 2023 ([19]) |
| Giredestrant (GDC-9545) | Oral SERD | ER+, HER2– advanced BC (in trials) | Oral capsule, Phase 3 (Roche) |
| Vepdegestrant (Veppanu) | Oral PROTAC (ER degrader) | ER+, HER2–, ESR1-mutated advanced BC after ≥1 ET | Oral tablet, approved May 2026 ([1]) ([2]) |
| Tamoxifen | SERM (estrogen receptor modulator) | Hormone receptor–positive breast cancer (adjuvant or metastatic) | Oral, long history, generic (1977 FDA) |
| Letrozole, Anastrozole, etc. | Aromatase inhibitors (ER ligand deprivation) | Postmenopausal ER+ breast cancer (adjuvant or metastatic) | Oral, long history |
Notes: Therapies are grouped by general class. All approved use in metastatic ER+/HER2– disease unless noted. ESR1-mutant indication is shown in bold. (Sources: FDA labels and press releases ([19]) ([1]) ([2]) ([48]).)
Table 2. VERITAC-2 Phase 3 Results in ESR1-Mutant Patients
| Endpoint | Vepdegestrant (Veppanu) | Fulvestrant (control) | Hazard Ratio (95% CI) |
|---|---|---|---|
| Number of ESR1-mutant patients | 270 | 270 | – |
| Median PFS (months) | 5.0 (95% CI 3.7–7.4) | 2.1 (95% CI 1.9–3.5) | 0.57 (0.42–0.77); p<0.0001 ([3]) ([4]) |
| Confirmed Response Rate (ORR, %) | 19% (95% CI 12–27%) | 4% (95% CI 1.6–10%) | (–) |
| Disease Control Rate (CR+PR+SD, %) | Not reported | Not reported | – |
| Estimated 6-month PFS rate (%) | Data not reported | Data not reported | – |
| Median OS (months) | Not reached (16% events) | Not reached | NA |
Results from the pre-specified analysis of ESR1-mutant subgroup. Vepdegestrant significantly prolonged PFS vs. fulvestrant by blinded central review ([3]) ([4]). Overall survival was immature at analysis. Response assessments by RECIST 1.1 ([3]).
6. Case Studies and Expert Perspectives
Although widespread clinical experience with vepdegestrant is new, several practitioners involved in the pivotal trial have highlighted its promise. Dr. Erika Hamilton, principal investigator of VERITAC-2, emphasized that Veppanu gives clinicians “another tool in the breast cancer treatment arsenal” and “brings renewed hope” for ESR1-mutant patients ([12]). Her comments illustrate the patient-facing impact: many women with metastatic ER+ disease have run through standard therapies; an effective oral agent with a new mechanism can improve quality of life and delay disease progression in some of these cases. Similarly, Arvinas’s CMO praised that the approval addresses “an aggressive form of breast cancer” and meets an “unmet need” ([31]). In future case series or real-world studies, expect reports of patients with refractory ESR1-mutant tumors achieving partial remission or stable disease on Veppanu for several months when nothing else worked; these anecdotal cases will complement the trial statistics.
From an academic oncology perspective, Veppanu validates years of preclinical modeling. Early trials of ARV-471 in the neoadjuvant setting (OSPREY/VERITAC-1 studies) showed robust ER knockdown in tumors, establishing proof-of-mechanism ([40]). Oncologists and researchers will be eager to see follow-up data on circulating ESR1-mutant clones during therapy, durability of ER degradation, and any patterns of acquired resistance. For example, does long-term use select for tumors that upregulate E3 ligase regulators or AR pathway cross-talk? These are important research questions now that PROTAC is clinically validated.
From an investment and biotech viewpoint, this approval signals a paradigm shift. As noted in industry analyses, the targeted protein degradation field had been poised for commercialization in 2026 ([55]). The first clinical validation tends to launch a flurry of dealmaking and funding. After Veppanu’s announcement, both Kymera and C4 Therapeutics saw changes in stock price, and new collaborations were already underway (e.g. Gilead–Kymera) ([52]). Analysts are looking ahead to the successes of analogous programs – for example, the CELMoD (cereblon E3 ligase modulator) products iberdomide and CC-220 from BMS and Judd, or AR degraders in prostate – as further confirmation of the modality’s viability ([49]) ([55]).
In summary, multidisciplinary perspectives converge: oncologists are cautiously optimistic about the new option for ESR1-mutant patients ([12]); researchers see validation of protein degradation science ([8]); and industry stakeholders view this as a watershed that could replicate the success story of antibody-drug conjugates (another modality that required decades of science before blockbuster drugs emerged ([55])).
7. Future Directions and Conclusions
The FDA approval of vepdegestrant in May 2026 is a major milestone that is rippling across oncology and drug development. It likely heralds an era in which protein degradation – previously an intriguing concept – becomes a mainstream therapeutic approach. Multiple companies now have “first-in-class” programs chasing other targets. The PROTAC paradigm will be rigorously tested in the coming years across indications.
Key short-term developments to watch include:
- Other PROTAC Approvals: Given Veppanu’s success, similar PROTACs may enter pivotal trials or regulatory review soon. Arvinas’s AR degraders in prostate (ARV-110/ARV-766) could be the next candidates; in fact, ARV-110 is in Phase 3 for CRPC. Meanwhile, early mover Nurix has an oral PROTAC BTK degrader (NX-2127) in trials, and others like C4’s IKZF degrader should report data. Positive phase 3 readouts (like Roche’s giredestrant or Amgen’s SERDs) will also be compared and contrasted to PROTAC performance.
- Combination Strategies: Vepdegestrant trials in combination (VERITAC-3, combinations with CDK4/6 inhibitors, PI3K inhibitors, etc.) are planned ([71]). These studies will examine whether PROTACs can enhance standard regimens. If PROTAC combinations show synergy, that opens more uses beyond the ESR1-mutant niche.
- Biomarker Development: ESR1 mutation testing was built into this approval. Future protein degrader approvals may also require companion diagnostics (e.g. to detect the target or pathway activation). We may see more integrated genomic testing in oncology practice.
- New Targets: PROTAC research is venturing into areas beyond nuclear receptors. For example, the Aurora Kinase A inhibitor alisertib has failed in the clinic but a new PROTAC against Aurora A could revive it. Similar stories exist with others (cyclin E/Cdk2, STAT family, YAP/TAZ, etc.). Companies are also exploring chimeric platforms targeting the lysosome or autophagy (LYTACs, AUTACs, etc.) to degrade extracellular and filamentous targets, expanding the “protein degrader” concept even further.
Long-term, if PROTACs consistently demonstrate benefit, one can imagine a shift in pharmacology education and regulatory science: the traditional focus on receptor occupancy may give way to measuring protein knockdown. Pharmaceutical pipelines will likely keep a mix of inhibitors and degraders, choosing the best tool for each target. Companies that have built novel drug platforms (Arvinas with PROTACs, Monte Rosa with AI-driven glues, Nurix with CRBN-based degraders, etc.) may enjoy valuation premiums. Indeed, an investment report noted that right now (2025–2026) is “the best time to invest in targeted protein degradation,” highlighting leading assets like Arvinas, BMS, Kymera, Nurix, C4T, etc ([72]).
In conclusion, the FDA’s May 2026 approval of vevdegestrant (Veppanu) is not just a new drug label but a “proof of principle” for a whole drug class. It validates decades of biochemical research and opens the door to a flood of new degraders. For ER+ breast cancer patients with ESR1 mutations, it provides a desperately needed new option ([11]). For the biotechnology field, it reaffirms that proteasomal degradation is a powerful modality. The “protein degrader pipeline” – already spanning oncology, immunology, neurology and more – now has a clinical anchor. Time will tell whether future PROTACs achieve the lofty hopes placed upon them, but with vepdegestrant’s success, the journey from the bench to the bedside has truly begun.
References: As cited above, all claims and data in this report are drawn from peer-reviewed publications, regulatory filings, press releases, and expert analyses ([1]) ([3]) ([7]) ([4]) ([6]) ([49]) ([73]) ([58]) (full citations are included in the embedded links).
External Sources (73)

Need Expert Guidance on This Topic?
Let's discuss how IntuitionLabs can help you navigate the challenges covered in this article.
I'm Adrien Laurent, Founder & CEO of IntuitionLabs. With 25+ years of experience in enterprise software development, I specialize in creating custom AI solutions for the pharmaceutical and life science industries.
DISCLAIMER
The information contained in this document is provided for educational and informational purposes only. We make no representations or warranties of any kind, express or implied, about the completeness, accuracy, reliability, suitability, or availability of the information contained herein. Any reliance you place on such information is strictly at your own risk. In no event will IntuitionLabs.ai or its representatives be liable for any loss or damage including without limitation, indirect or consequential loss or damage, or any loss or damage whatsoever arising from the use of information presented in this document. This document may contain content generated with the assistance of artificial intelligence technologies. AI-generated content may contain errors, omissions, or inaccuracies. Readers are advised to independently verify any critical information before acting upon it. All product names, logos, brands, trademarks, and registered trademarks mentioned in this document are the property of their respective owners. All company, product, and service names used in this document are for identification purposes only. Use of these names, logos, trademarks, and brands does not imply endorsement by the respective trademark holders. IntuitionLabs.ai is an AI software development company specializing in helping life-science companies implement and leverage artificial intelligence solutions. Founded in 2023 by Adrien Laurent and based in San Jose, California. This document does not constitute professional or legal advice. For specific guidance related to your business needs, please consult with appropriate qualified professionals.
Related Articles

Datroway FDA Approval for First-Line Metastatic TNBC
Review the FDA approval of Datroway (datopotamab deruxtecan) for first-line metastatic TNBC. Analyze TROPION-Breast02 efficacy, survival, and safety data.

Relacorilant: FDA Approved GR Antagonist for Ovarian Cancer
Review the FDA approval of relacorilant (Lifyorli) for platinum-resistant ovarian cancer. Learn its clinical efficacy and mechanism as a GR antagonist.

Biomarker Testing Coordination Services: An Oncology Guide
Updated 2026 guide to biomarker testing coordination services in oncology. Covers navigator roles, FDA companion diagnostic reclassification, liquid biopsy adoption, NCCN guideline updates, and AI-driven diagnostics for precision medicine.